Galactose-1-phosphate uridyl transferase deficiency; Galactokinase deficiency; Galactose-6-phosphate epimerase deficiency; GALT; GALK; GALE; Epimerase deficiency galactosemia; GALE deficiency; Galactosemia type III; UDP-galactose-4; Duarte variant DefinitionGalactosemia is a condition in which the body is unable to use (metabolize) the simple sugar galactose. CausesGalactosemia is an inherited disorder. This means it is passed down through families. If both parents carry a nonworking copy of the gene that can cause galactosemia, each of their children has a 25% (1 in 4) chance of being affected with it. This is called autosomal recessive inheritance. There are 3 forms of the disease:
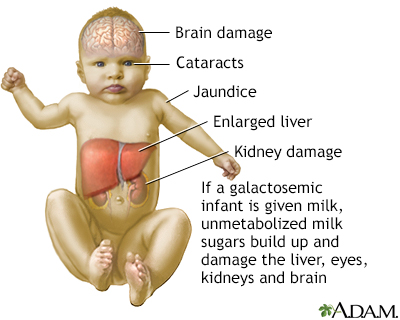
People with galactosemia are unable to fully break down the simple sugar galactose. Galactose makes up one half of lactose, the sugar found in milk. If an infant with galactosemia is given milk, substances made from galactose build up in the infant's system. These substances damage the liver, brain, kidneys, and eyes. People with galactosemia cannot tolerate any form of milk (human or animal). They must be careful about eating other foods containing galactose. SymptomsInfants with galactosemia may show symptoms in the first few days of life if they eat formula or breast milk that contains lactose. They may develop a serious blood infection with the bacteria E coli. Symptoms of galactosemia are:
Exams and TestsTests to check for galactosemia include:
In many states, newborn screening tests check for galactosemia. Test results may show:
TreatmentPeople with this condition must avoid all milk, products that contain milk (including dry milk), and other foods that contain galactose, for life. Read product labels to make sure you or your child with the condition are not eating foods that contain galactose. Infants can be fed:
Calcium supplements are recommended. Support GroupsMore information and support for people with galactosemia and their families can be found at: Galactosemia Foundation -- www.galactosemia.org Outlook (Prognosis)People who are diagnosed early and strictly avoid milk products and other foods that contain lactose can live a relatively normal life. However, mild mental impairment may develop, even in people who avoid galactose. Possible ComplicationsThese complications can develop:
When to Contact a Medical ProfessionalContact your health care provider if:
PreventionIt is helpful to know your family history. If you have a family history of galactosemia and want to have children, genetic counseling will help you make decisions about pregnancy and prenatal testing. Once the diagnosis of galactosemia is made, genetic counseling is recommended for other members of the family. Many states screen all newborns for galactosemia. If the newborn test shows possible galactosemia, they should contact the child's provider right away for advice about giving their infant milk products. They should also ask the provider about having blood tests that can be done to confirm a diagnosis of galactosemia. ReferencesBerry GT. Classic galactosemia and clinical variant galactosemia. 2000 Feb 4 [updated 2021 Mar 11]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews [Internet]. Seattle, WA: University of Washington. PMID: 20301691 pubmed.ncbi.nlm.nih.gov/20301691/. Bonnardeaux A, Bichet DG. Inherited disorders of the renal tubule. In: Yu ASL, Chertow GM, Luyckx VA, Marsden PA, Skorecki K, Taal MW, eds. Brenner and Rector's The Kidney. 11th ed. Philadelphia, PA: Elsevier; 2020:chap 44. Hijazi G, Kishnani PS. Defects in metabolism of carbohydrates. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 107. Pearl PL, DiBacco ML, Gibson KM. Inborn errors of metabolism and the nervous system. In: Jankovic J, Mazziotta JC, Pomeroy SL, Newman NJ, eds. Bradley and Daroff's Neurology in Clinical Practice. 8th ed. Philadelphia, PA: Elsevier; 2022:chap 91. | |
| |
Review Date: 4/8/2025 Reviewed By: Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. The information provided herein should not be used during any medical emergency or for the diagnosis or treatment of any medical condition. A licensed medical professional should be consulted for diagnosis and treatment of any and all medical conditions. Links to other sites are provided for information only -- they do not constitute endorsements of those other sites. No warranty of any kind, either expressed or implied, is made as to the accuracy, reliability, timeliness, or correctness of any translations made by a third-party service of the information provided herein into any other language. © 1997- A.D.A.M., a business unit of Ebix, Inc. Any duplication or distribution of the information contained herein is strictly prohibited. | |
| |

 Galactosemia
Galactosemia